T²F-Pharm
Pharmacophore models are an accurate and minimal tridimensional abstraction of intermolecular interactions between chemical structures, usually derived from a group of molecules or from a ligand-target complex. Only a limited amount of solutions exists to model comprehensive pharmacophores using the information of a particular target structure without knowledge of any binding ligand. In the T²F-Pharm project, we developed an automated and customable tool for truly target-focused pharmacophore modeling.
Key molecular interaction fields of a macromolecular structure are calculated using the AutoGRID energy functions. The most relevant points are selected by a newly developed filtering cascade and clustered to pharmacophore features with a density-based algorithm. This method represents an extremely valuable instrument for drug design in a situation of scarce ligand information available, but also in the case of underexplored therapeutic targets, as well as to investigate protein allosteric pockets and protein-protein interactions.
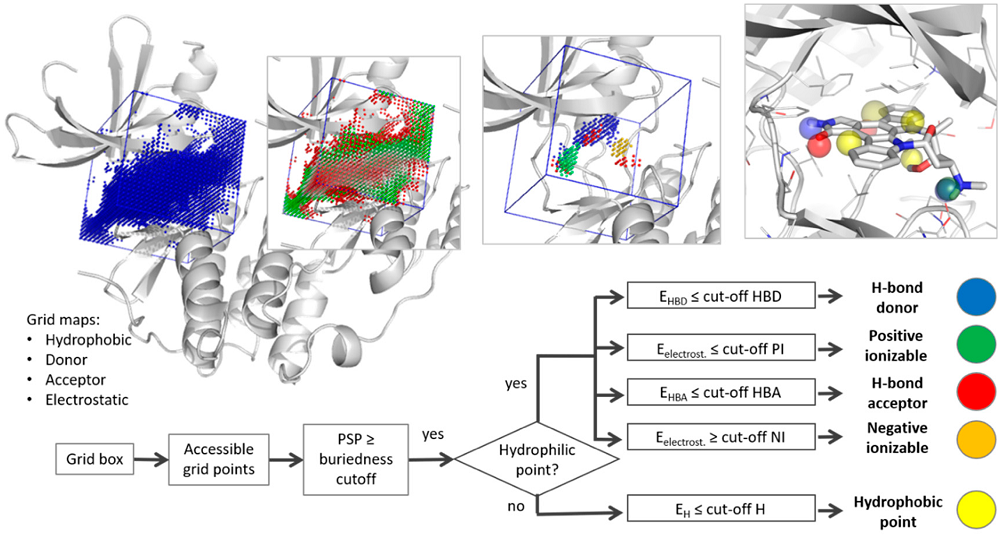
Figure: T²F-Pharm allows to generate pharmacophore models from apo binding sites by extracting energy hot spots for important pharmacophoric features (figure taken from Mortier, 2018).
People
Funding
- Bundesministerium für Bildung und Forschung, grant ID 031A262C
Publications
- (2018). Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces. Molecules 23(8):1959. DOI: 10.3390/molecules23081959.